Sickle Cell Anemia Curable Via Nutrition
(Click Link) http://organicbodycleanse.com/wp-content/uploads/2015/11/Sickle-Cell-Cure.pdf
 Sickle Cell Anemia Curable Via Nutrition
Sickle Cell Anemia Curable Via Nutrition
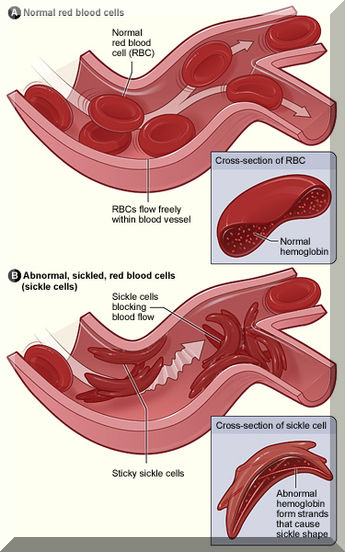
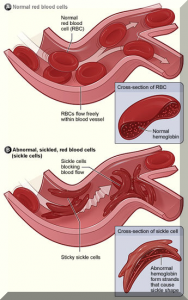
Sickle-cell disease (SCD), also known as sickle-cell anaemia (SCA) and drepanocytosis, is a hereditary blood disorder, characterized by an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells.
This leads to a propensity for the cells to assume an abnormal, rigid, sickle-like shape under certain circumstances. Sickle-cell disease is associated with a number of acute and chronic health problems, such as severe infections, attacks of severe pain (“sickle-cell crisis”), and stroke, and there is an increased risk of death.
Sickle-cell disease occurs when a person inherits two abnormal copies of the haemoglobin gene, one from each parent. Several subtypes exist, depending on the exact mutation in each haemoglobin gene. A person with a single abnormal copy does not experience symptoms and is said to have sickle-cell trait. Such people are also referred to as carriers.
The complications of sickle-cell disease can be prevented to a large extent with vaccination, preventive antibiotics, blood transfusion, and the drug hydroxyurea/hydroxycarbamide. A small proportion of patients require a transplant of bone marrow cells.
Almost 300,000 children are born with a form of sickle-cell disease every year, mostly in sub-Saharan Africa, but also in other parts of the world such as the West Indies and in people of African origin elsewhere in the world. In 2013 it resulted in 176,000 deaths up from 113,000 deaths in 1990.
The condition was first described in the medical literature by the American physician James B. Herrick in 1910, and in the 1940s and 1950s contributions by Nobel prize-winner Linus Pauling made it the first disease where the exact genetic and molecular defect was elucidated.
ref: https://en.wikipedia.org/wiki/Sickle-cell_disease
(Click Link) Sickle Cell Anemia Curable Via Nutrition





